


臨床試験成績
国内第Ⅲ相臨床試験(FSN-011-01P-02試験)
富士製薬工業株式会社社内資料: 子宮を有する日本人更年期障害女性におけるオープン試験[承認時評価資料]
エフメノ®カプセル100mg 有効性と安全性(解説動画)
試験概要
| 目的 | HRTの施行対象である、子宮を有し、更年期障害又は卵巣欠落症状を有する日本人女性を対象とし、エストロゲン製剤に本剤を併用した際の子宮内膜増殖症の発症抑制効果と安全性を検討する。 |
|---|---|
| 試験デザイン | 多施設共同、非無作為化、オープン試験 |
| 対象 | 子宮を有し、更年期障害又は卵巣欠落症状を有する日本人女性349例 |
| 試験方法 |
エストラジオール1日1回0.5又は1.0mg経口投与の併用下で、本剤を1周期(28日)の1~28日目まで1日1回100mgを就寝前に経口投与(持続的投与法)、又は1周期(28日)の15~28日目まで1日1回200mgを就寝前に経口投与(周期的投与法)した。投与期間はいずれも52週間とし、後観察期間を4週間とした。 
|
| 評価項目 |
(1)有効性 (2)安全性 |
| 解析計画 |
解析対象集団最大の解析対象集団(FAS):345例 有効性の解析主要評価項目:子宮内膜増殖症の発現率とClopper-Pearson型信頼区間を算出した。本剤投与時の子宮内膜増殖症発現率の許容限界値は、日本人更年期障害又は卵巣欠落症状を有する患者を対象に実施されたエストラジオール・酢酸ノルエチステロン経皮吸収型製剤の国内第Ⅲ相臨床試験、並びに海外の主要ガイドラインであるCHMPガイドラインを参考にし、投与後1年間での子宮内膜増殖症発現率の両側95%信頼区間の上限が2.0%を下回ることを確認する計画とした。主要評価項目(子宮内膜増殖症の発現率)に関しては、子宮内膜の病理判定に係る中央判定委員会の最終判定結果において「陽性:子宮内膜増殖症(単純型、複雑型)、細胞異型を伴う子宮内膜増殖症、異型ポリープ状腺筋腫、がん」と判定されたものとした。 主要評価項目の副次解析として投与方法別(持続的投与法、周期的投与法)の事前に規定されたサブグループ解析を行った。 副次評価項目:経腟超音波検査による子宮内膜の厚さを評価時点ごとに、測定値と、その測定値のベースラインからの変化量について、投与方法別と全体で算出した 安全性の解析本剤又は併用薬(エストラジオール)の投与開始日から最終観察終了日までの間に発現したものを有害事象として取り扱った。 |
患者背景
人口統計学的特性及び他の基準値の特性(連続量データ)(FAS)
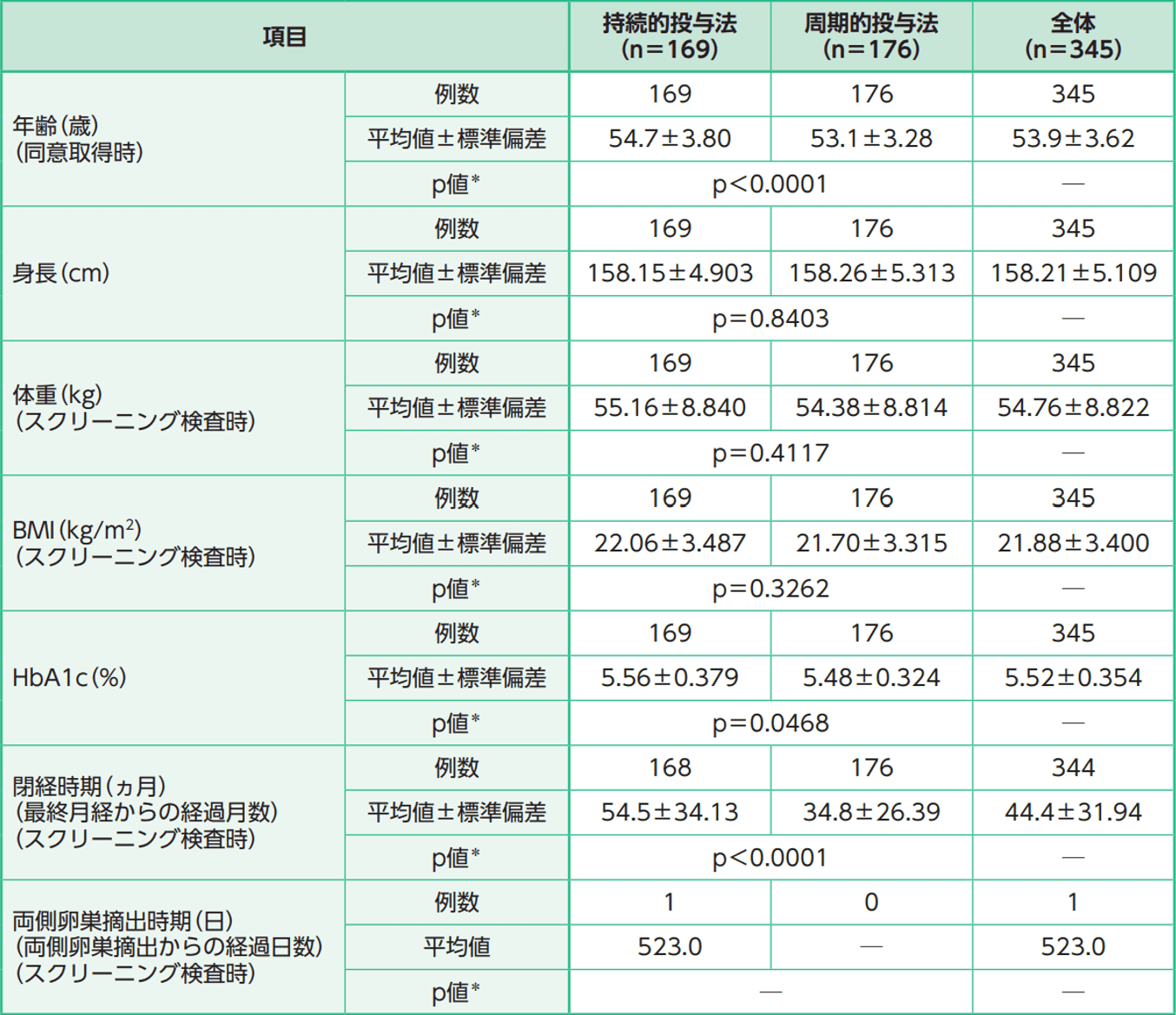
*: 投与方法間のt検定(Studentのt検定)
参考:患者背景
参考:人口統計学的特性及び他の基準値の特性(カテゴリカルデータ)(FAS)
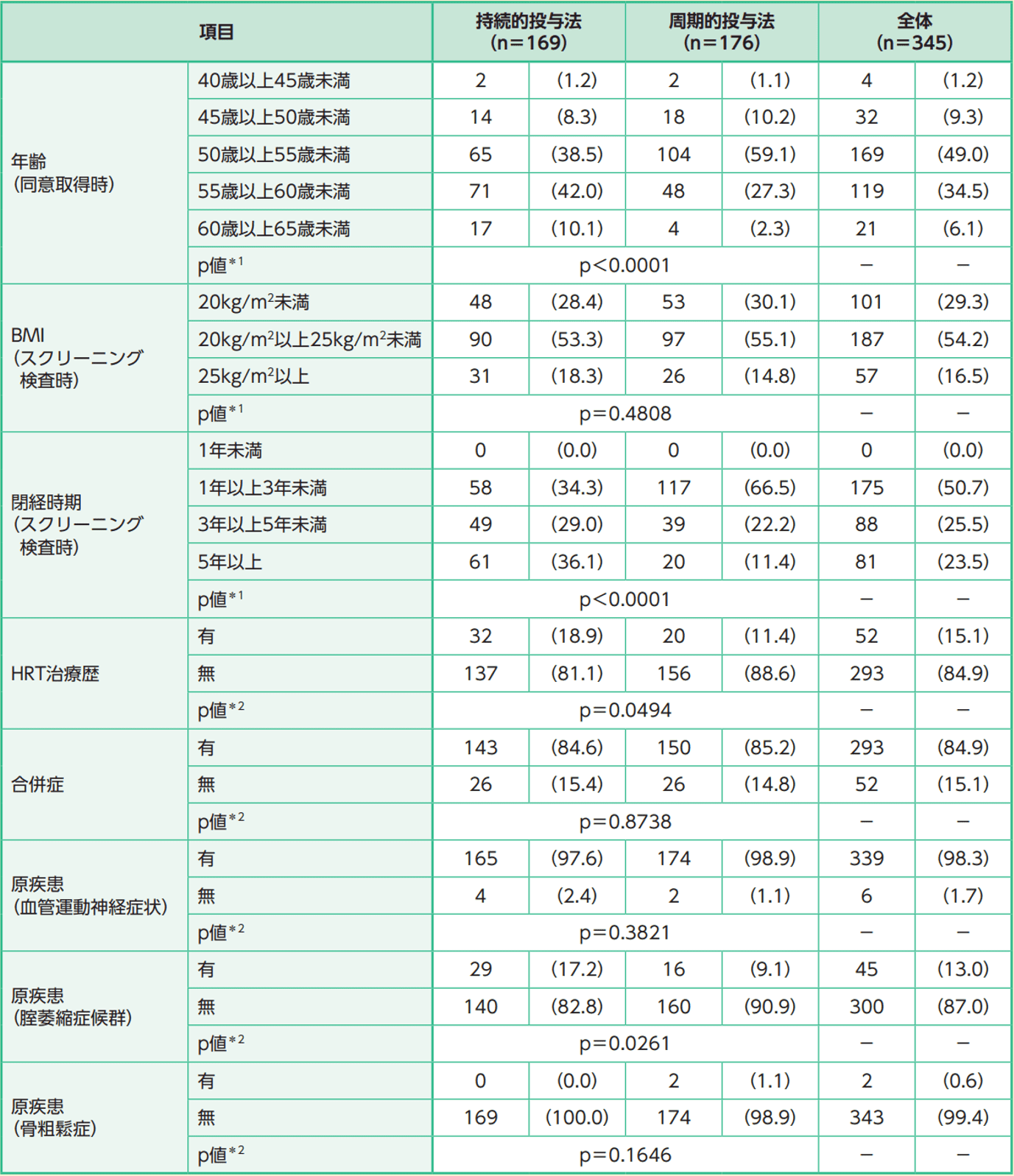
例数(%)
*1: 投与方法間のWilcoxon順位和検定
*2: 投与方法間のχ2検定
子宮内膜増殖症の発現率(主要評価項目)
<主解析;検証的な解析結果>
FAS345例中、17例は主要評価項目のデータが得られず、7例は主要評価項目の評価データ検査を基準期間内に実施しなかったため、主要評価項目の対象症例は321例であった。判定結果除外となった3例を除いた318例を解析したところ、主要評価項目である子宮内膜増殖症の発現率は0.0%(0/318例)であった。また、両側95%信頼区間は0.00−1.15%であり、95%信頼区間の上限が事前に定めた水準である2.0%を下回ったことが確認された。
子宮内膜増殖症の発現率(52週又は中止時)(FAS)
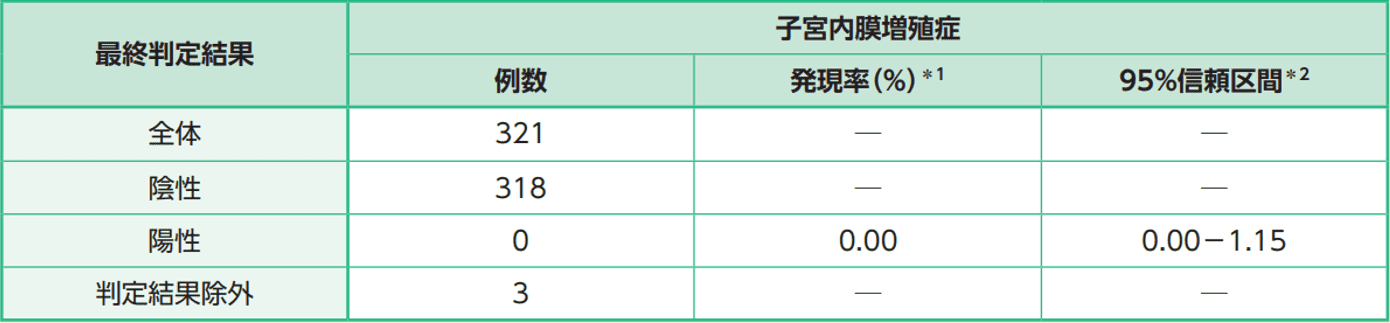
*1:分母=(陰性+陽性)
*2:Clopper-Pearson型信頼区間
<サブグループ解析>
投与方法別の子宮内膜増殖症の発現率は持続的投与法で0.0%(0/156例)、周期的投与法で0.0%(0/162例)であった。
子宮内膜増殖症の発現率(投与方法別、サブグループ解析)(52週又は中止時)(FAS)
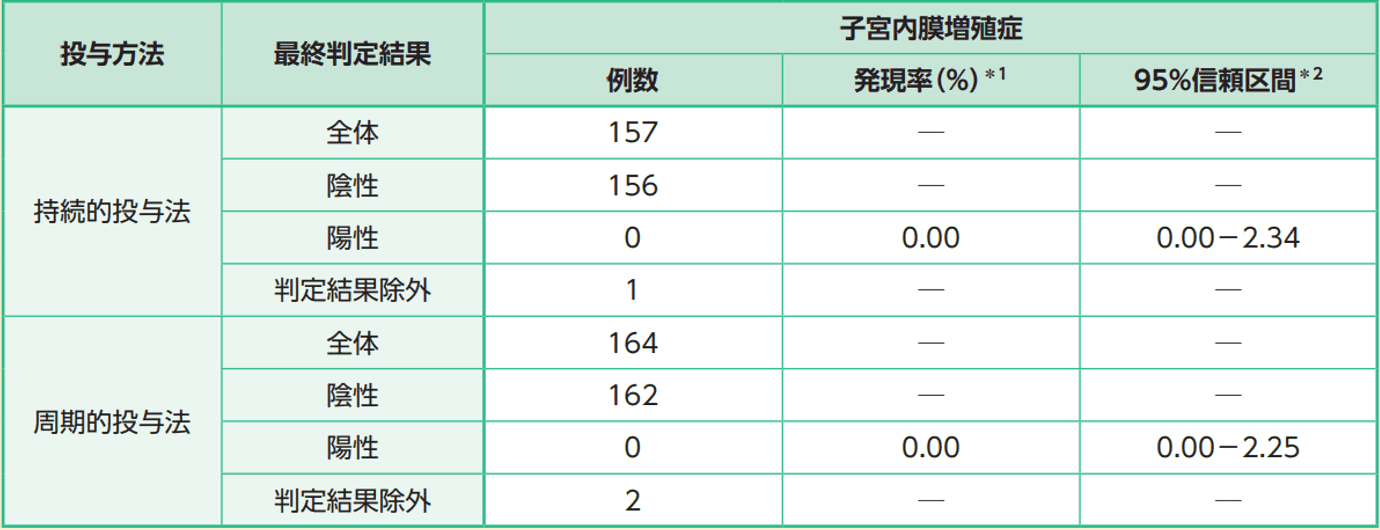
*1:分母=(陰性+陽性)
*2:Clopper-Pearson型信頼区間
投薬期間中の子宮内膜の厚さ(副次評価項目)
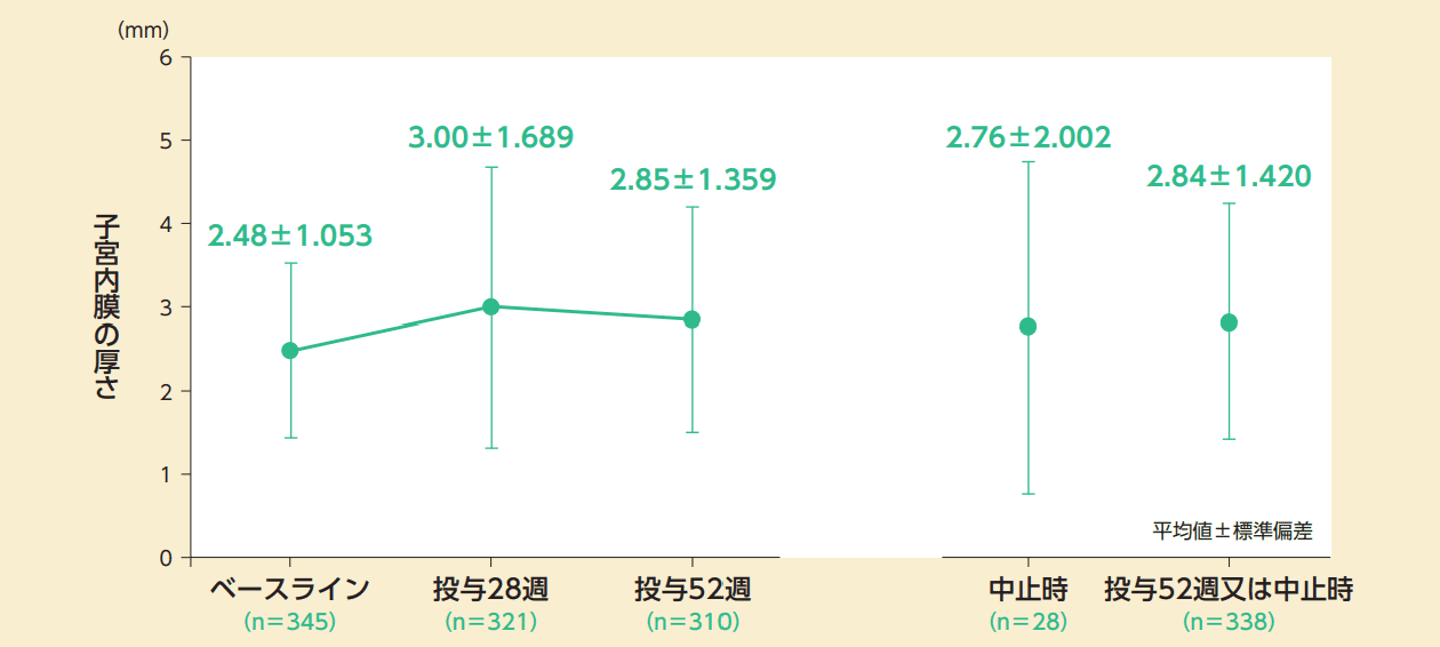
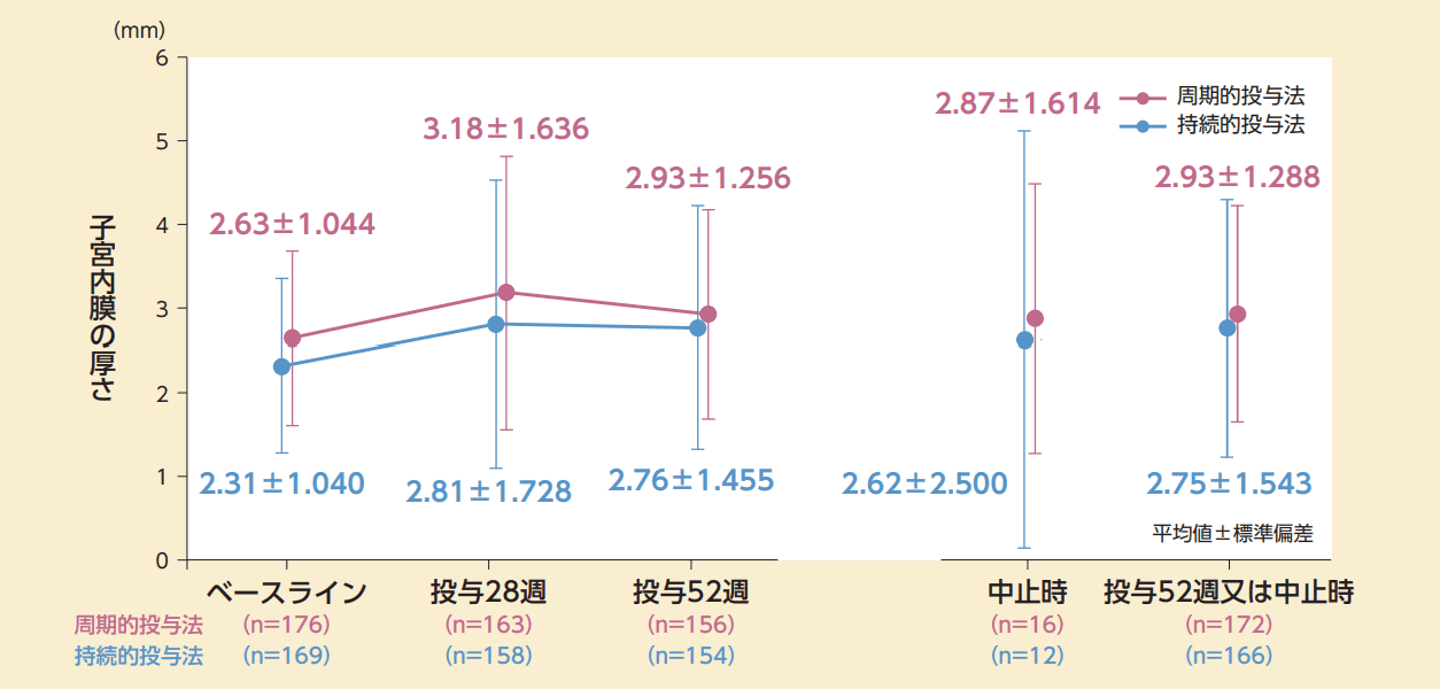
副次評価項目である評価時点ごとの子宮内膜の厚さの推移は以下のとおりであった。52週又は中止時の子宮内膜の厚さのベースラインからの変化量(平均値±標準偏差)は持続的投与法で0.43±1.363mm、周期的投与法で0.32±1.248mmであった。
⼦宮内膜の厚さの推移(FAS)
子宮内膜の厚さの推移(投与方法別、サブグループ解析)(FAS)
安全性
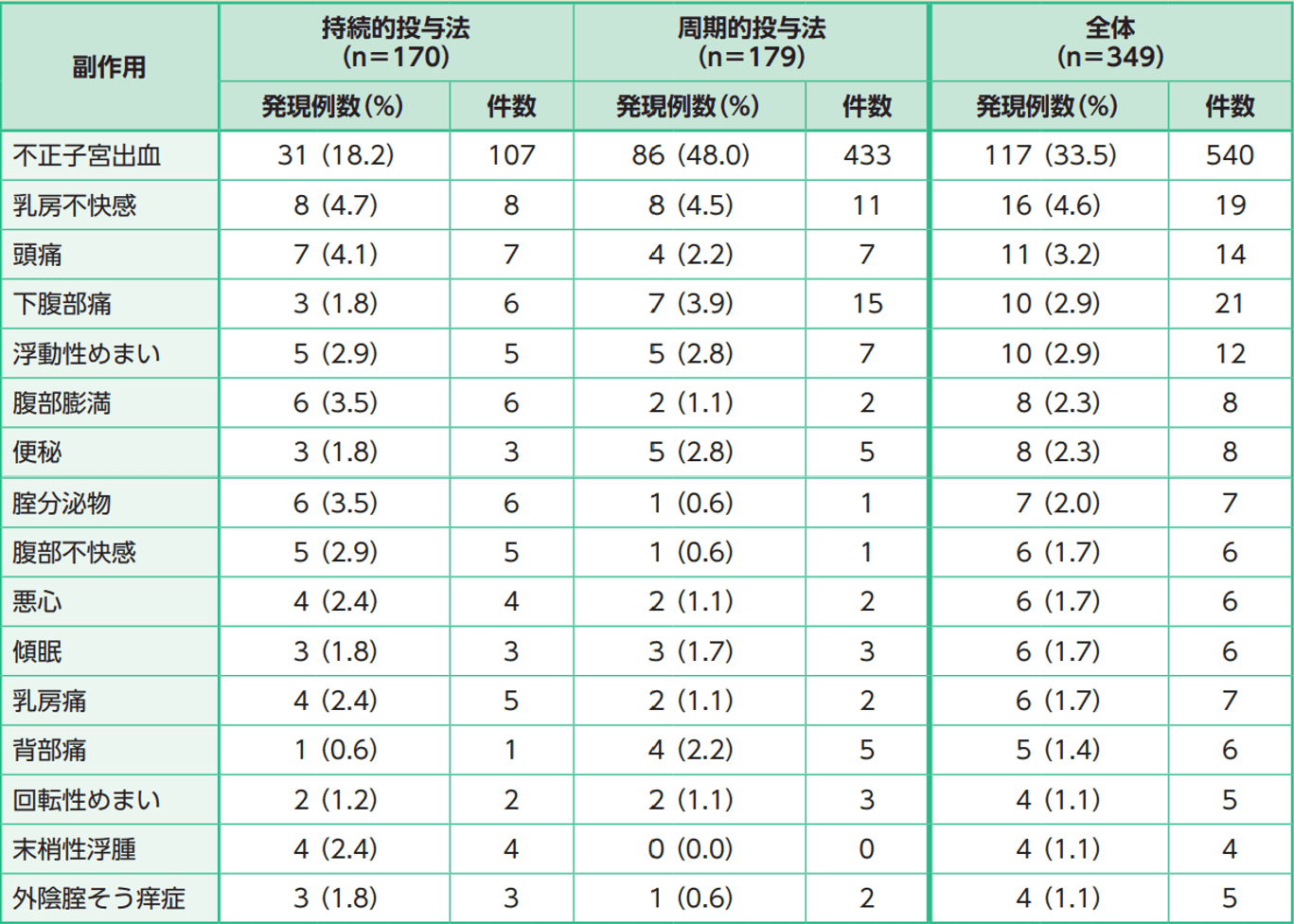
副作用は持続的投与法で49.4%(84/170例)、周期的投与法で58.7%(105/179例)、全体で54.2%(189/349例)に発現した。主な副作用は、不正子宮出血が33.5%(117/349例)、乳房不快感が4.6%(16/349例)、頭痛が3.2%(11/349例)、下腹部痛、浮動性めまいが各2.9%(10/349例)、腹部膨満、便秘が各2.3%(8/349例)、腟分泌物が2.0%(7/349例)等であった。
本試験において、死亡に至った副作用の発現は認められなかった。重篤な副作用は2例2件であった。内訳は、乳腺浸潤性小葉癌及び乳管内増殖性病変でいずれも持続的投与法であった。投与中止に至った副作用は全体で7例11件(持続的投与法で5例7件、周期的投与法で2例4件)発現した。中止に至った副作用で2例以上に発現した事象は頭痛(2例)で、いずれも持続的投与法であった。
主な副作用の発現状況(発現率1%以上)(SAS)
MedDRA/J Ver.23.0
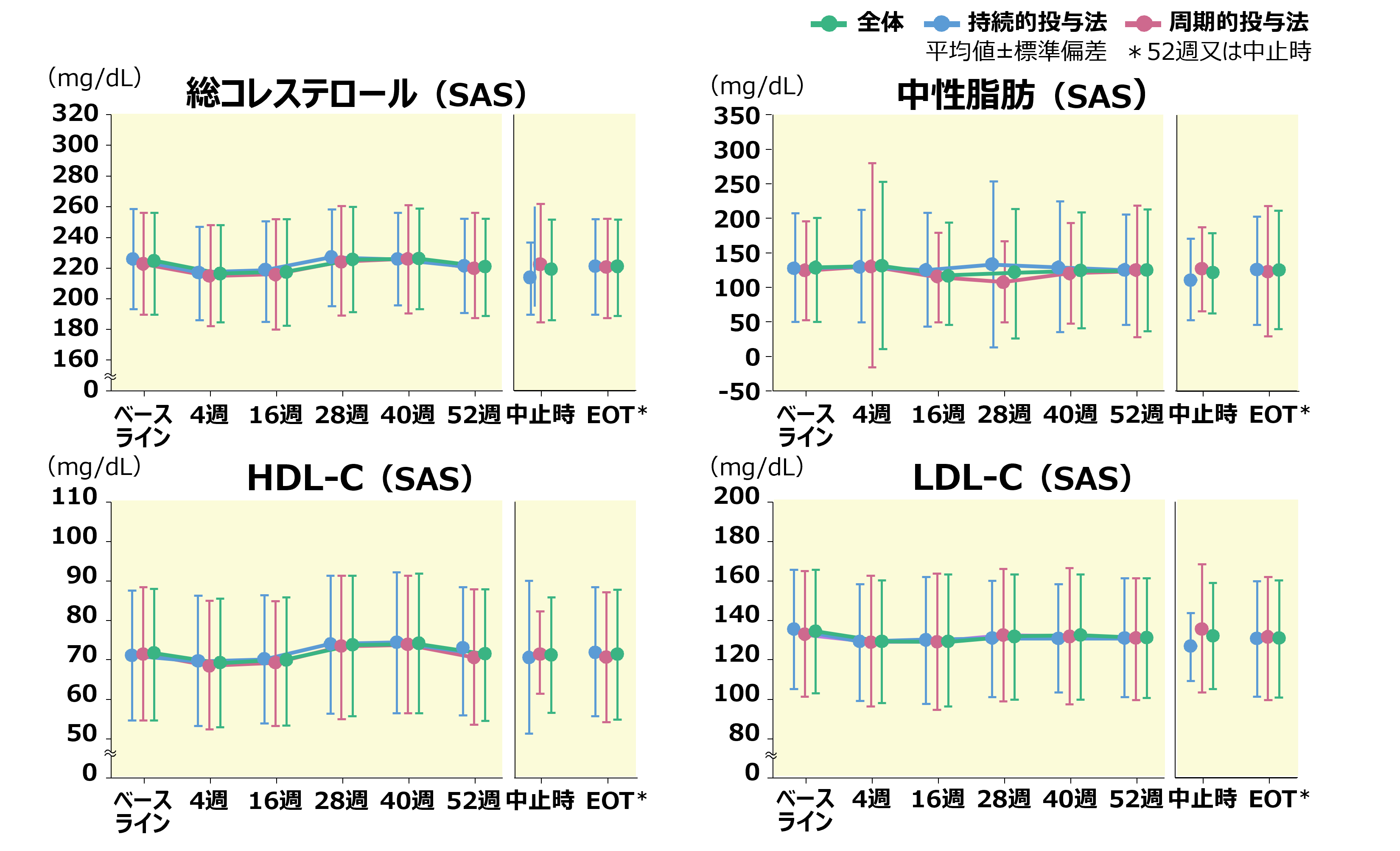
臨床検査値の推移(投与方法別、サブグループ解析)
富士製薬工業株式会社社内資料:子宮を有する日本人更年期障害女性におけるオープン試験[承認時評価資料]
海外第Ⅳ相臨床試験(PEPI 1995、1996試験)(海外データ)
The Writing Group for the PEPI Trial: JAMA. 1995; 273(3): 199-208.[PMID:7807658]
富士製薬工業株式会社社内資料: 海外臨床試験(PEPI 1995, 1996)[承認時参考資料]
The Writing Group for the PEPI Trial: JAMA. 1996; 275(5): 370-375.[PMID:8569016]
試験概要
| 目的 | 外国人閉経後女性を対象とし、プラセボ、結合型エストロゲン(CEE)単独、CEE+プロゲストーゲンの投与を行った際の、子宮内膜の組織学的所見を検討する。 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 試験デザイン | 多施設共同、無作為化、プラセボ対照、二重盲検試験 | ||||||||||
| 対象 | 閉経後女性875例(子宮を有する女性:596例、子宮を摘出した女性:279例) | ||||||||||
| 試験方法 |
以下の投与方法で行った。治療期間は3年間であった。
|
||||||||||
| 評価項目 |
(1)有効性 (2)安全性 |
||||||||||
| 解析計画 |
有効性の解析ベースライン特性についての治療レジメン間の差は分散分析及びFisherの正確確率検定で評価した。イベントの発現率は頻度及びパーセンテージで表示し、治療レジメン間の発現率の差はFisherの正確確率検定で評価した。 安全性の解析主要評価項目についてはF検定によるnominal p値をBonferroni法により上方調整し、p<0.05の時にt検定を行った。推定効果の95%信頼区間はこの調整なしで算出した。 |
子宮内膜評価
CEE+プロゲストーゲンの投与を行った3群では単純型(嚢胞性)増殖症が10例、複雑型(腺腫性)増殖症が2例、異型増殖症が1例発現した。異常の発現率にプラセボ群との有意な差は認められなかった。(p=0.16、Fisherの正確確率検定;名目上のp値)
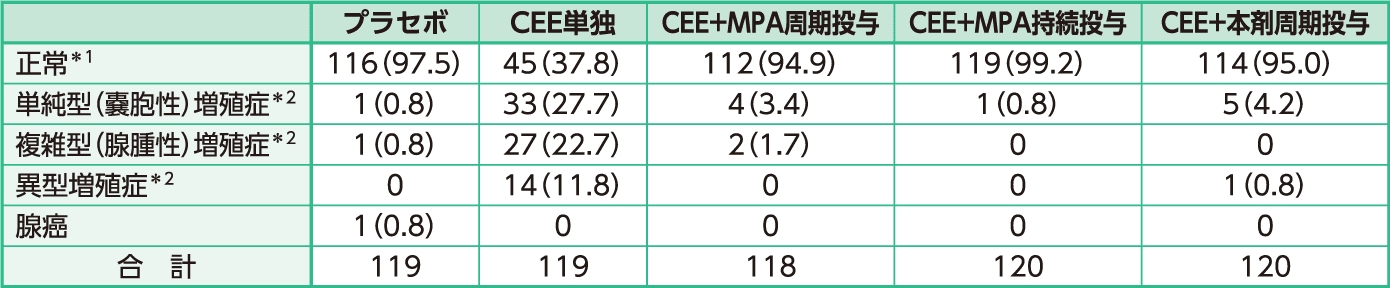
例数(%)
病理学者3名の判定が異なり、婦人科医が採用した判定を用いた30件を含む。
*1:p=0.16(正常 vs 異常) プラセボ vs CEE+MPA(周期)、CEE+MPA(持続)、CEE+本剤(Fisherの正確確率検定;名目上のp値)
*2:p<0.001 プラセボ vs CEE単独(Fisherの正確確率検定;名目上のp値)
フォローアップ時の主要評価項目のベースラインからの平均変化量及び共分散分析結果(検証的結果)
HDL-C及びフィブリノゲンで有意な群間差が認められた(いずれもBonferroni p<0.001、F検定)。CEE単独群とCEE+本剤周期投与群のHDL-Cの平均増加量はそれぞれ5.6、4.1mg/dLであり、CEE+MPA周期投与群とCEE+MPA持続投与群に比べ、有意に大きかった(Bonferroni p<0.004、t検定)。フィブリノゲンの変化は実薬群に比べプラセボ群で有意な増加が認められた(全てのBonferroni p≦0.02、t検定)。実薬群間には有意差は認められなかった(t検定)。収縮期血圧及び平均インスリンレベル(食後2時間値)には群間に有意差は認められなかった(F検定)。
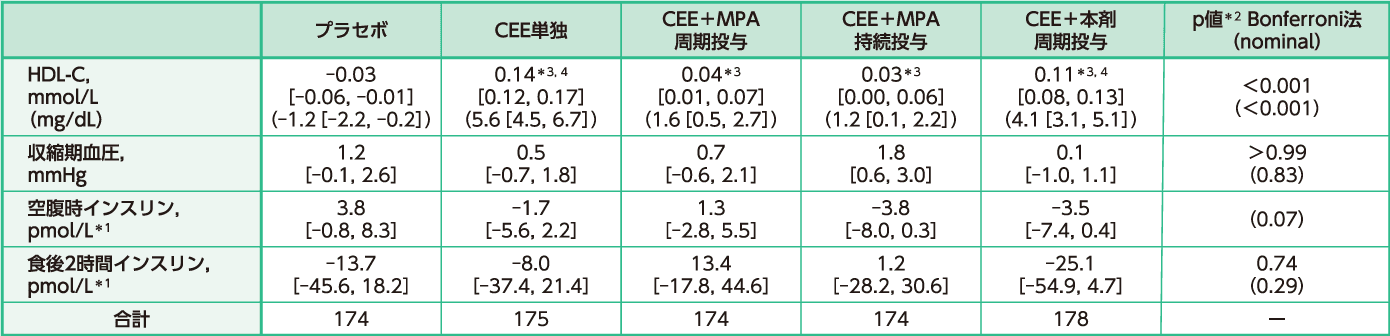
ベースラインからの平均変化量[95%信頼区間]
共変量:施設、子宮摘出の有無、ベースライン値(コホート間でベースライン値に差が見られた場合)
※:フィブリノゲン変化量について、CEE単独群のベースラインからの平均変化量と95%信頼区間の値の文献中の数値に矛盾があり、正しい数値の確認ができなかったことからデータは掲載できなかった。
*1:対数変換データから逆変換
*2:F検定
*3:p<0.001(vs プラセボ)(Bonferroni, t検定)
*4:p<0.004(vs CEE+MPA)(Bonferroni, t検定)
有害事象
試験期間中、97例110件の有害事象が発現し、プラセボ群で8例10件、CEE単独群で47例57件、CEE+MPA周期投与群で18例18件、CEE+MPA持続投与群で8例8件、CEE+本剤周期投与群で16例17件であった。
その内訳は、プラセボ群でその他の癌*、子宮内膜増殖症、胆嚢疾患、子宮摘出術が各2件、子宮内膜癌、乳癌が各1件、CEE単独群で子宮内膜増殖症が41件、子宮摘出術が7件、血栓塞栓症が4件、胆嚢疾患が2件、子宮内膜癌、乳癌、心血管疾患が各1件、CEE+MPA周期投与群でその他の癌*、胆嚢疾患が各4件、子宮摘出術が3件、乳癌、血栓塞栓症、子宮内膜増殖症が各2件、心血管疾患が1件、CEE+MPA持続投与群で胆嚢疾患が5件、血栓塞栓症が2件、その他の癌*が1件、CEE+本剤周期投与群で乳癌、胆嚢疾患が各4件、心血管疾患が3件、血栓塞栓症、子宮摘出術が各2件、その他の癌*、子宮内膜増殖症が各1件であった。
乳癌が発現した8例のうち、2例(CEE+本剤周期投与群及びCEE+MPA周期投与群各1例)は無作為化から6ヵ月から1年の期間で発現した。別の3例(CEE+本剤周期投与群が2例、CEE+MPA周期投与群が1例)は1年目の検査で乳癌が診断された。
本試験において死亡は3例であり、その内訳は乳癌(CEE+本剤周期投与群)、肝癌(CEE+本剤周期投与群)、肺癌(CEE+MPA周期投与群)によるものが各1例であった。
ホルモン補充療法の中止が必要であったのは、エストロゲン依存性と考えられる癌、脳卒中、一過性脳虚血発作、肺塞栓症、深部静脈血栓症、腺腫性増殖症、異型増殖症、心停止、心筋梗塞及びその他の癌*であった。
* 子宮内膜癌、乳癌、非黒色腫皮膚がん以外
※ MPAは「更年期障害及び卵巣欠落症状に対する卵胞ホルモン剤投与時の子宮内膜増殖症の発症抑制」の効能又は効果に対して国内未承認
本剤の効能又は効果、用法及び用量は以下の通りである。
4. 効能又は効果
更年期障害及び卵巣欠落症状に対する卵胞ホルモン剤投与時の子宮内膜増殖症の発症抑制
6. 用法及び用量
卵胞ホルモン剤との併用において、以下のいずれかを選択する。
- 卵胞ホルモン剤の投与開始日からプロゲステロンとして100mgを1日1回就寝前に経口投与する。
- 卵胞ホルモン剤の投与開始日を1日目として、卵胞ホルモン剤の投与15日目から28日目までプロゲステロンとして200mgを1日1回就寝前に経口投与する。これを1周期とし、以後この周期を繰り返す。
国内第Ⅰ相臨床試験
血中濃度
単回投与
健康閉経後成人女性12例に本剤200mgを絶食時に単回経口投与したときの薬物動態パラメータは以下のとおりである。
血清中プロゲステロン濃度の推移
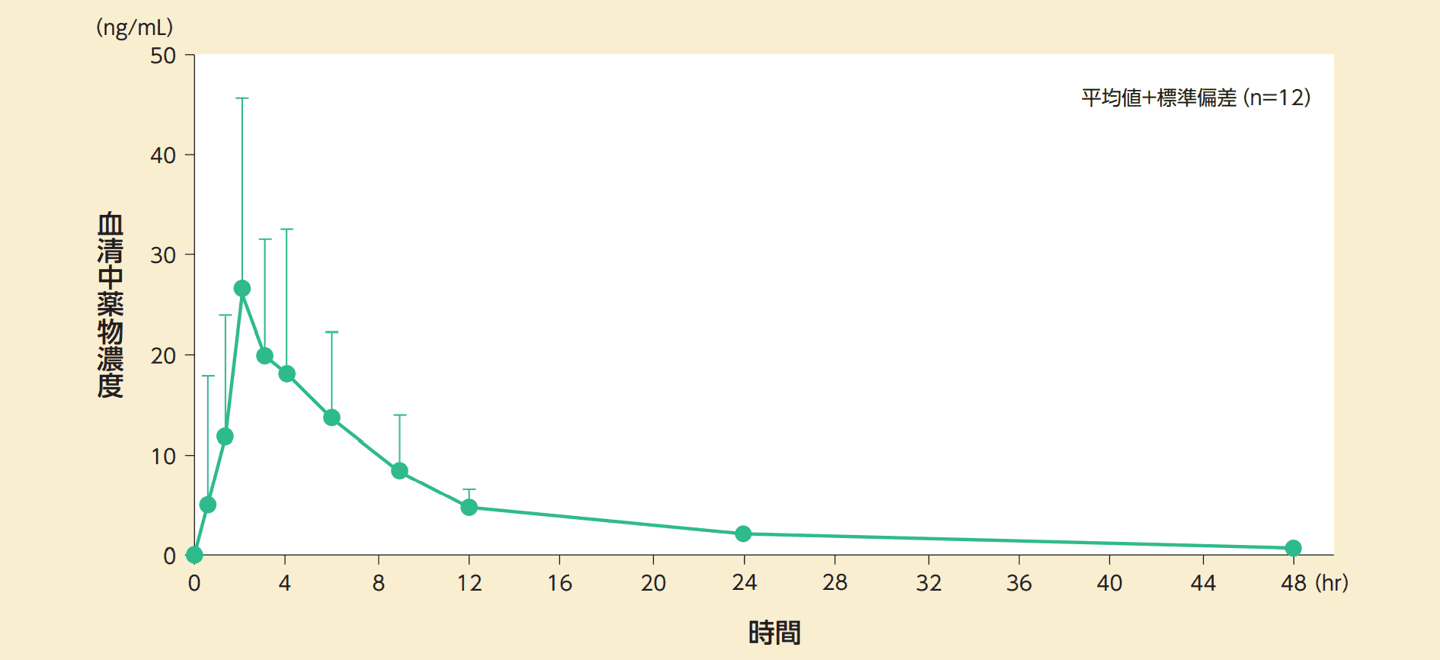
単回投与時の薬物動態パラメータ

平均値±標準偏差
富士製薬工業株式会社社内資料: 健康閉経後女性における薬物動態試験(単回投与及び食事の影響)
反復投与
健康閉経後成人女性計20例に本剤100mg又は200mgを絶食時に1日1回7日間反復経口投与した。Cmax及びAUCはいずれも100mg投与群と200mg投与群で用量依存性を示した。本剤100mg又は200mgを1日1回反復経口投与したときの薬物動態パラメータは以下のとおりである。
血清中プロゲステロン濃度の推移
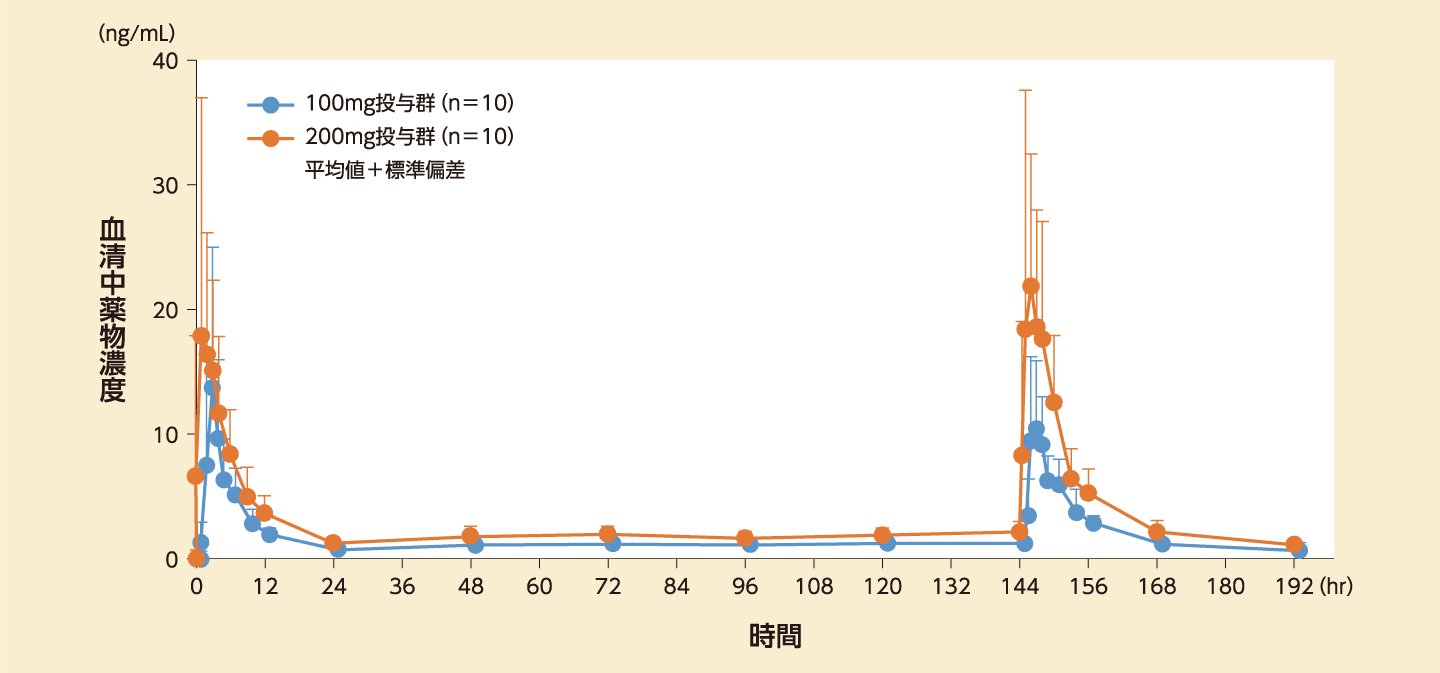
反復投与時(1回100mg/日又は200mg/日)の薬物動態パラメータ
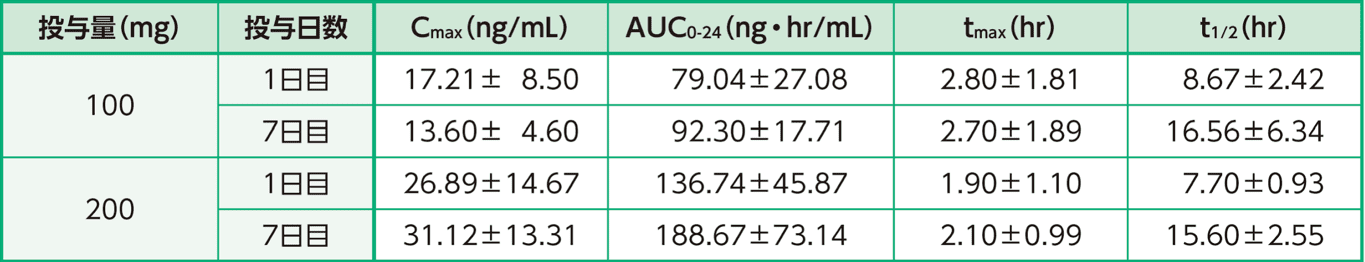
平均値±標準偏差
富士製薬工業株式会社社内資料: 健康閉経後女性における薬物動態試験(反復投与)
食事の影響
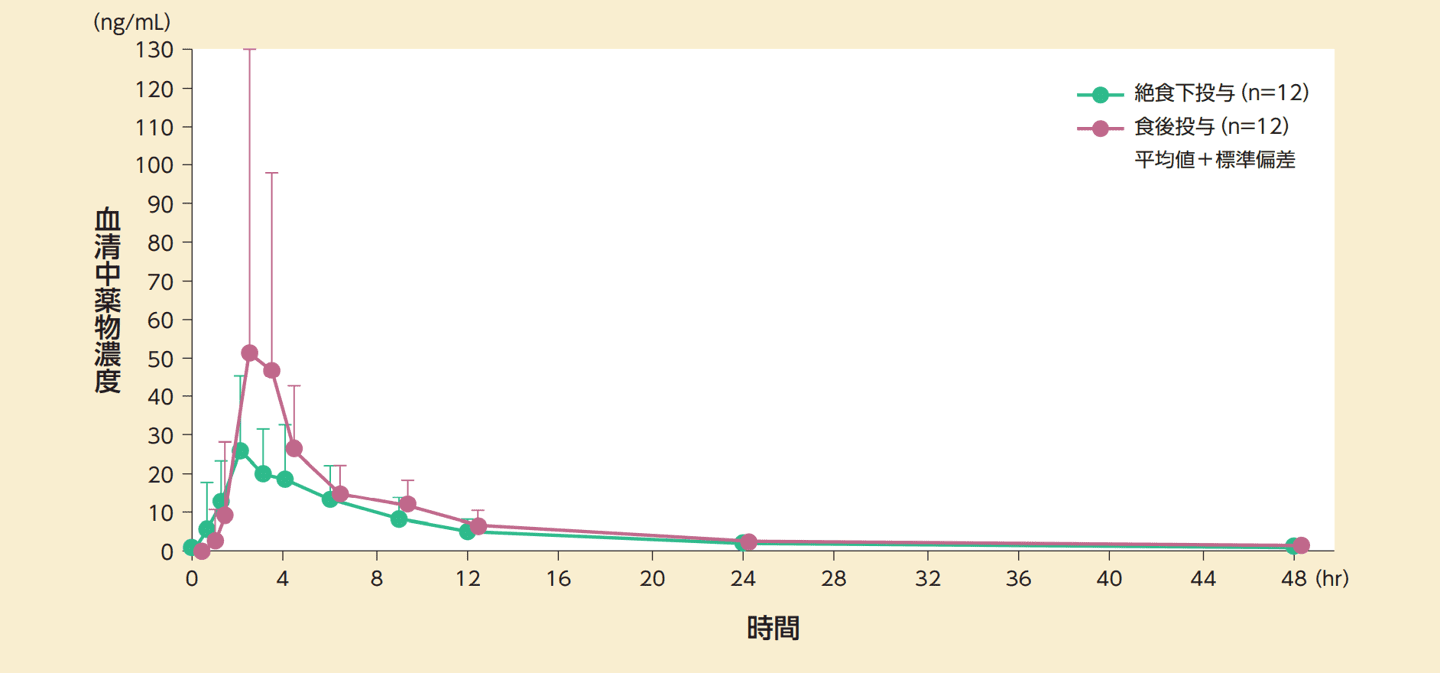
健康閉経後成人女性12例に、本剤200mgを絶食時及び食後に単回経口投与したとき、AUC及びCmaxは絶食時投与に比べ食後投与で上昇する傾向が示された。また、tmax及びt1/2は食事の影響を受けないことが示された。
血清中プロゲステロン濃度の推移
絶食下及び食後の薬物動態パラメータ
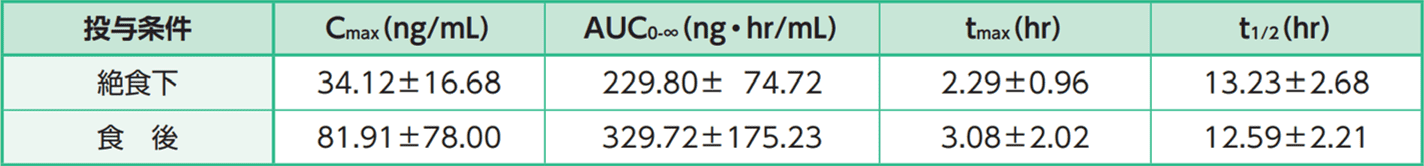
平均値±標準偏差
富士製薬工業株式会社社内資料: 健康閉経後女性における薬物動態試験(単回投与及び食事の影響)
6. 用法及び用量
卵胞ホルモン剤との併用において、以下のいずれかを選択する
- 卵胞ホルモン剤の投与開始日からプロゲステロンとして100mgを1日1回就寝前に経口投与する。
- 卵胞ホルモン剤の投与開始日を1日目として、卵胞ホルモン剤の投与15日目から28日目までプロゲステロンとして200mgを1日1回就寝前に経口投与する。これを1周期とし、以後この周期を繰り返す。
7. 用法及び用量に関連する注意
食後に本剤を投与した場合、Cmax及びAUCが上昇するとの報告がある。食事の影響を避けるため、食後の服用は避けること。